
Let’s Go to the Videotape: Promoting the visibility and understanding of regulatory science by using case studies from publicly available FDA information

Regulatory Science and the Need for Education/Training
Almost 10 years ago, the United States Food and Drug Administration (FDA) recognized the need to stimulate the pace of innovation in drug and biologics development when it issued its 2006 report on “Innovation/Stagnation—Challenge and Opportunity on the Critical Path to New Medical Products”.
At the time, the pharmaceutical industry was struggling with unsuccessful product developments that were either failing to produce marketable products or unable to take advantage of recent advances in science, such as in bioinformatics, genomics, and genetic mapping of diseases. The FDA recognized that something had to be done to capture the attention of those in the field of drug development and regulation, because the problem belonged to everyone and the solutions could not come only from the government, industry, academia, or other constituencies alone.
This FDA report set the stage for regulatory science as the centerpiece of translational science. While the report was broad in scope and multidisciplinary in subject matter, some aspects were of interest to biostatisticians, including how they might contribute (O’Neill, 2006).
The Critical Path report was a call for a broad constituency to take better advantage of the technologies and scientific advances available to develop and bring medical products to patients in a more timely and efficient manner. The report also acknowledged a pressing need for scientists who understand the scientific regulatory process; the scientific standards of evidence that medical products are held to; and how everything from discovery through clinical trials, safety monitoring, and life-cycle benefit/risk assessment are carried out.
This need is relevant to recruiting and FDA scientists, including external scientific and clinical advisors who sit on the FDA’s advisory committees; pharmaceutical industry scientists who currently are trained on the job in a relatively inefficient process of trial and error; and academics who develop course material and are charged with developing the next generation of scientists and clinical investigators in drug development science.
More specifically, the Critical Path report addressed the need to advance the field of regulatory science, since there are virtually no programs at the university graduate level in this topic area. A number of academic programs focus on the field of regulatory affairs, but that field should not be confused with regulatory science.
In 2011, the Institute of Medicine held a special workshop and issued a report on strengthening the workforce in regulatory science. In the same year, the FDA issued “Advancing Regulatory Science at FDA”, its Strategic Plan for Regulatory Science, in which the FDA defined regulatory science as the science of developing new tools, standards, and approaches to assess the safety, efficacy, quality, and performance of FDA-regulated products.
That report identified eight priority areas of regulatory science where new or enhanced engagement is essential to the continued success of the agency’s public health and regulatory mission. These priority areas are:
- 1. Modernize Toxicology to Enhance Product Safety
- 2. Stimulate Innovation in Clinical Evaluations and Personalized Medicine to Improve Product Development and Patient Outcomes
- 3. Support New Approaches to Improve Product Manufacturing and Quality
- 4. Ensure FDA Readiness to Evaluate Innovative Emerging Technologies
- 5. Harness Diverse Data through Information Sciences to Improve Health Outcomes
- 6. Implement a New Prevention-focused Food Safety System to Protect Public Health
- 7. Facilitate Development of Medical Countermeasures to Protect against Threats to U.S. and Global Health and Security
- 8. Strengthen Social and Behavioral Science to Help Consumers and Professionals Make Informed Decisions about Regulated Products
The science of statistics, and of regulatory statistics in particular, is a critical component of regulatory decision making. The field of statistics, as it is applied and used in pharmaceutical product regulation, has been maturing over the past 40 years or so, with the explosion of new journals devoted to one or more aspects of statistical planning, data evaluation, and decision making in drug development and regulation. Yet, few, if any, graduate-level statistical programs or courses are devoted to the field of regulatory statistics.
A New Course in Regulatory Science
To begin to address this need, we developed and presented a course at the Harvard T. H. Chan School of Public Health called Statistical and Quantitative Methods for Pharmaceutical Regulatory Science, which we now have given annually during the eight-week spring quarter semesters of 2014, 2015, and 2016. The goal of this course is to enable scientists and public health professionals who already have an introductory background to biostatistics and clinical trials to acquire the competencies in quantitative skills and systems thinking required to understand and participate in drug development and regulatory review processes.
The course illustrates, from a regulatory perspective, how statistical and quantitative methods are used to transform information into evidence demonstrating the safety, efficacy, and effectiveness of drugs and therapeutic biologicals over the course of a medical product’s life cycle. The course is also intended to illustrate how evidence is evaluated and weighed over the life cycle of a pharmaceutical product, since regulatory science is very much about evaluating and weighing evidence from a scientific perspective. The public does not currently have the opportunity to be exposed to what this actually means in practice.
In addition to lectures developed by one of the authors based upon his experiences at the FDA, the course includes and is structured around materials that are publicly available on the FDA’s website and in case studies from the FDA’s public advisory committee archives. One goal is to inform others about what FDA materials are publicly available, as well as emphasize the statistical component of regulatory decision making and make materials available to a wider audience, including academics, that can be used for educating and training the next generation of scientists interested in medical product development and regulation.
This article describes the course content and design, including its use of video clips of FDA advisory committees and some of the considerations that went into the design of the course, such as decisions about the intended audiences. We also look at its main objectives, designed so the course would not compete with nor be a substitute for other university subject-matter courses, such as clinical trials, epidemiology, statistical methods, pharmacometrics, data mining, or benefit/risk evaluations. On the contrary, we designed this course primarily to illustrate how statistical thinking is incorporated into regulatory decision making. It was further developed to be a prototype and first in a series of courses that might eventually lead to a graduate degree program in regulatory science.
The course is not focused on statistics at a deep technical level, despite the fact that many of the statistical methodological issues faced by the FDA are complex and often without precedent or a clear consensus on their use for decision making. The course is intended to be of interest to a broader audience, including clinical investigators and post-graduate scientists who are early in their careers, but have some experience in drug development science. That audience includes many of the clinical and scientific staff who are employed by university hospitals and biotechnology and pharmaceutical firms in the Boston area.
While the course tested the feasibility of learning objectives built around regulatory science topics and case studies, it also strived to engage and stimulate other academics and industry scientists to develop similar materials and course programs with use of the publicly available information that is available on the FDA’s website.
Content of the Course
The course consists of 16 two-hour lectures, each with a brief 10-minute break in the middle, delivered twice per week over eight weeks.
The first lecture of the course presents an “Overview of the Evolution of Evidentiary Standards for Efficacy and Safety of Drugs and Biologics: the FDA product life cycle review and evaluation process.” The material traces how the FDA evidentiary standards changed or were modified over the years, and how the design and analysis of clinical studies has improved as a consequence of these evolving standards and the regulatory review process for implementing the standards.
The second lecture, “The Product Review Life Cycle—a brief overview,” provides students with a high-level view and understanding of how the pre-market and post-market stages of drug development, evaluation, and review fit together in the regulatory environment of life-cycle evaluation of medical products. Subsequent lectures present more specific attention to the later stages.
Week 2 includes two lectures on the “Investigational Drug Review Process” that describe how the FDA provides advice and evaluation of industry clinical trial protocols, mainly dealing with critique and advice on clinical trial design, and sponsor’s choice of clinical studies before they are conducted. Since this process is usually confidential with a sponsor, the public is not normally exposed to the issues encountered in the planning stages of development.
The purpose of this content is to illustrate the types of advice that FDA reviewers provide to sponsors about a proposed study’s ability to meet evidentiary standards and to support labeling indications.
As part of these lectures, we typically introduce a publicly available case study on design or choice, or a methodology-based approach from or based upon a video clip of a real-life FDA advisory committee session.
During the third week, two lectures focusing on the “New Drug Application (NDA) Review Process” describe how FDA reviewers evaluate completed studies and evidence submitted by a pharmaceutical company seeking marketing approval. These lectures provide an opportunity to introduce examples from actual FDA advisory committees to illustrate real-world examples on various topics of interest.
Week 4 covers several quantitative aspects of safety assessment in the product life-cycle evaluation, with a specific focus on randomized clinical trials that have been evaluated by the FDA during the pre-market stage. Case studies from the FDA advisory committees illustrate safety evaluation issues and give students an understanding of how advisory committee members evaluate evidence.
Week 5 lectures cover a high-visibility safety topic that spanned multiple years and multiple advisory committee meetings: a case study that involves the drug Avandia (rosiglitazone), a thiazolidinedione-class medication used in the treatment of diabetes mellitus type 2, and its association with cardiovascular risk. This example is important because it involved evaluation of the relative evidentiary weight of randomized trials, observational studies, meta-analysis of observational and randomized trials, and the precedent-setting introduction of a pre-market requirement to rule out cardiovascular risk of a certain magnitude.
Week 6 topics include issues of post-marketing and post-approval safety assessment, emphasizing the role of voluntary reporting, data mining for safety signal detection, the newly developed FDA Sentinel system for active surveillance using electronic health claims records, and a case study of an anti-coagulant drug (Pradaxa®—dabigitran) evaluated over its life cycle for safety concerns.
In Week 7, there are two lectures on the unique challenges in the use and acceptance of foreign clinical data for regulatory decision making, with emphasis on International Conference for Harmonization (ICH) guidance on regulatory acceptance of evidence from foreign clinical studies and the increasing use of the multi-regional clinical trial.
During Week 8, we bring in other academic scientists to discuss, in panel format, their experiences as advisory committee members and members of data monitoring committees. This session gives students an opportunity to interact with scientists who have real-world experience in the field. We invite Harvard faculty who have served as advisory committee members to serve on these panels. Among those participating have been L.J. Wei, Michael Hughes, Scott Evans, Sharon Lise-Normand, David Schoenfeld, Richard Gelber, Cyrus Mehta, and Ralph D’Agostino.
Depending on the constraints of an eight-week course, many other topics can be considered that are relevant to regulatory statistics, including unique study designs for biomarker evaluation and use in confirmatory trials, other population-enrichment and adaptive designs that prune potentially winning treatments, non-inferiority designs, the evaluation of central auditing strategies in cancer clinical trials, and the impact and consequences of missing data in clinical trials. Some of these lectures can be modeled around FDA guidance on a specific topic.
A by-product of the course:
Showcase the unique role of independent FDA advisory committee members and the use of advisory committee-based case studies to educate/train potential new members before they attend a first meeting.
In a recent article in this publication, Michael Proshan (2016) shared his experiences as a biostatistician on an FDA advisory committee and the unique role he played as an independent, non-FDA scientist in providing objective advice to the FDA on complex evidentiary issues.
FDA advisory committee members are usually academics or other government scientists who are vetted for conflicts of interest and provide independent advice to the FDA on scientific matters of evidence brought to the committees. In any one year, 150 external scientists and clinicians are on the 14 advisory committees of the Center for Drug Evaluation and Research (CDER) and serve in this role. These scientists and clinicians may not be fully familiar with FDA standards of evidence or with the dynamics of a publicly held advisory committee, and the challenge of evaluating evidence presented by both FDA and a sponsor to address FDA questions regarding meeting the evidentiary standards.
The course videos derived from the FDA advisory committees serve as a rich source of training and educational materials for future advisory committee members, and could be crafted solely for that purpose. Currently, these videos have not been used that way.
Motivation for Selecting Course Content
In deciding what to present in this course, we were motivated by the following goals.
Increase public understanding of the evidentiary framework in the United States for efficacy, safety, and continued market availability of drugs, biologics, etc.
If you ask most people about how the prescription drugs they take to treat an acute or a chronic condition or to prevent a future serious health event were tested to evaluate their claims of efficacy and safety, you would probably get the answer, “I don’t know.” If you were to ask many health scientists, clinicians, or university scientists the same question, the answers might be somewhat different, but are likely to be vague. If you were to ask that question of a biostatistician or a statistician in the health sector, you might get different answers from a clinical trial investigator, an epidemiologist, a statistical or mathematical modeler, a data analyst, or a scientist in the pharmaceutical product development space.
Each of these scientists is working in a singular space without knowledge of how it all fits together. In fact, were you to address that question to most faculty members in schools of public health, medical schools, schools of pharmacy, or departments in the arts and sciences, the answers probably would not be particularly useful in informing the course content that they teach or the students they mentor.
This is a problem and a challenge for the educational system, as well as for all stakeholders interested in the development and availability of new therapies to promote public health. It is surprising that little, if any, graduate-level course material deals with the scientific aspects of medical product development and regulation, especially with regard to the roles that biostatistics theory, methods, and principles play.
Challenge the educational enterprise in training scientists in regulatory science
Drug discovery, development, regulation, and surveillance come together to create a team sport that involves many disciplines in education. To a large extent, clinical investigators are usually first trained in an academic setting with very little regulatory science exposure. These clinicians often find themselves working for pharmaceutical firms where that inexperience in regulatory science translates into less-than-optimal study design planning, safety evaluation strategies, or strategic planning that incorporates quantitative decision-making principles.
Currently, it appears that no discipline, school, or institution has responsibility for, let alone interest in, developing the emerging discipline of evidence development and evaluation to meet standards of regulatory approval. Current evidence development is a global enterprise, since most major pharmaceutical sponsors are multinational in scope and deal with multiple regulatory agencies around the globe. Essentially, we are now experiencing a new area of translational science and regulatory science, where most faculties have not had any experience or insight into how to craft new educational and training programs, if not courses, that would prepare their students to meet the challenges in this field.
The United States regulation of medical products is about the scientific assessment of evidence as it relates to the laws and regulations created by Congress to assure the safety and efficacy of marketed products. The process is complex, and based upon both legal and scientific standards that are increasingly more challenging and evolutionary to accommodate modern innovations, technology, and data collections strategies. These are all intended to provide evidence to support decision making about whether a medical product should be approved and made available to the public, or restricted in its use and availability to provide an adequate benefit-and-risk profile for the public and the individual who may take the product.
The growing interest in personalized medicine only raises the stakes that much higher, but the understanding of the complexities of this challenge appears weak at this time.
Address the question of what is the best educational medium to reach and benefit all the constituencies
Where in the educational enterprise can a student or professional be exposed to the core principles, processes, methods, tools, analysis strategies, study designs, active and passive surveillance approaches for safety assessment, and other emerging sciences supporting personalized medicine and better individualization of therapies to maximize benefit and minimize risk?
Currently, few programs exist to meet this challenge, and fewer courses have been developed for any of these topics, specifically focused on what is now called regulatory science. Of equal interest is whether in-class or online, on-demand formats would best serve the constituencies that would benefit from various educational and training efforts.
Current in-class educational models, even where lectures are videotaped and archived for repeat viewing, appear to fall short of optimizing access to information and class materials. Nor do current approaches provide face-to-face dialogue about the issues or integration of class assignments that expand the student’s level of understanding of the complexities of the problems. The current structure also does not encourage team interactions among diverse class population backgrounds. For the course developer, the challenge is to develop the materials at a level that is neither too simple for some and too challenging for others.
Determine the constituencies—the audience—and tailor content to them
The course we developed has now been given three times over three years to a graduate-level audience that has some basic understanding of statistical principles, but is generally too early in training or careers to benefit fully from the course material. The students have ranged from biostatisticians with master’s or PhD degrees to quantitatively oriented lawyers in a master’s in public health program to epidemiologists, clinical investigators, and other early industry-career types. Developing appropriate materials for this diverse audience is a challenge.
Consider the design of the course in statistical and quantitative decision making in pharmaceutical regulation
In the spring of 2014, the authors of this article collaborated to develop a course for the Harvard T. H. Chan School of Public Health that would expose graduate students to many aspects of assessing evidence of efficacy and safety of new drugs to satisfy the standards for approval in the United States.
The motivation for the course was to deal with topics that do not appear to be the subject of any current graduate programs. Considering that Boston is rapidly becoming a biotech hub for smaller, mid-size, and larger pharmaceutical firms, we thought there would be a need for some educational opportunity for university graduate students, professionals in these areas, and perhaps even faculty members to be exposed to what was occurring in a rapidly moving modern medical product development and regulation environment.
The other initial thinking was to open the course to different disciplines, not simply statisticians, who would be interested in how the various components of a complex and somewhat poorly known, and therefore poorly understood, process occurs.
Still further, we gave some consideration to creating a consortium to share the course content by inviting students from other universities in Boston who also might be interested and whose institutions did not offer programs covering the content we were planning to develop. This latter consideration has not materialized, due to the lead time needed and other coordination issues that had to be addressed.
Course Curriculum Structure and Evaluation
The evaluation of education curriculum must be based on a set of learning objectives and competencies. As stated in the syllabus, the overarching goal of this course is to enable scientists and professionals with a basic background in applied statistics to acquire the competencies in quantitative skills needed to contribute effectively to the drug development and regulatory review processes. The course focuses on providing an overview, awareness, and understanding of the important components of the regulatory review process with an emphasis on the statistical and quantitative aspects.
At the end of this first course, the student should be able to perform the following tasks:
- 1. Detail the characteristics of various stages of the life-cycle evaluation of a new drug.
- 2. Describe the required statistical and design aspects of the regulatory standards for evidence of efficacy and safety of a new drug.
- 3. Outline how the FDA’s implementation of regulations through science-based guidelines has shaped best practices.
- 4. Discuss how sponsors and the research community can cooperate in a framework of predictability for the scientific content needed for efficient pharmaceutical research.
- 5. Understand how substantial evidence of efficacy and safety is assessed and weighed throughout the pre- and post-approval stages of products.
- 6. Describe how safety considerations are monitored in the benefit and risk framework.
- 7. Explain how independent advice from external scientific advisors is used in the decision-making process.
To achieve these objectives, the course uses a blended learning approach involving lectures, web-based media, and selected case study examples derived from real-life FDA decision making and regulatory assessments that are available in the public domain to highlight and describe each phase of the regulatory drug approval process. The case studies illustrate regulatory science in action and practice, and include content publicly available from the FDA’s website that can be used in conjunction with FDA-science–based guidance and decision precedents.
Class sessions primarily introduce students to the areas described in these learning objectives and guide review and discussion of the case studies. Completion of web-based exercises, readings and videos, case study reviews, and assignments before and after the class lectures are critical for active participation and in-class discussions.
Class discussions and scheduled interactive case study sessions provide the opportunity for feedback, not only from the faculty, but from classmates.
The course web page and learning management system give students access to videotaped lectures, copies of the lecture slides, and web page links. In addition to the main course web page, there are five focused web pages: 1) Lecture Materials, 2) Lecture Videos, 3) Internet-based Resources, 4) Optional Educational Resources, and 5) Student Homework and Dropbox files.
As part of the course evaluation, each student is required to briefly describe and discuss their views of the cases presented. This interaction has been useful in helping students understand the statistical and regulatory issues, and the tasks undertaken by regulatory teams.
During the course, students acquired and refine their skills through class participation in the panel- and case-based studies discussions; critical evaluation of the provided regulatory guidances and published articles in the web-based references, readings, and videos; and assessment of design options in conducting drug development studies and implementing processes for reviewing the efficacy, safety, and effectiveness of programs by completing the required homework assignments.
The course grade is based upon class attendance and participation, enabling students to demonstrate that they are actively involved in reviewing and analyzing the readings and case studies. Class participation counts for 20% of the grade. Four individually prepared homework exercises, each counting for 20% of the grade, are required.
Each homework assignment consists of two major components: knowledge and practice. The knowledge component tests the student’s grasp and understanding of the presented material, often requiring the student to review the lecture slides or a taped lecture video. All lectures were videotaped and available in that format, along with the lecture slides, on demand for just this purpose.
The practice component consists of an exercise requiring that the student demonstrate skills in accessing new information to solve a decision-making or analytical reasoning problem not presented during the class session. Typically, each assignment included four to six such practice exercises. These are two examples of the practice-based examples from the first homework assignment.
- 1. Imagine that you are working for a pharmaceutical company in the United States and you want to advertise and promote a new prescription human drug directly to consumers through broadcast media, such as television and radio. Go to the appropriate webpage on the FDA website and find a guidance document that would help you understand what you might need to do. What is the name of the document and copy the document’s URL and paste below. Briefly summarize the major aspects of the document.
- 2. Go to the appropriate webpage on the FDA website and describe what serious safety issue needed further investigation in 9/2008 regarding the product Exubera,® an inhaled insulin product that was approved in 2006. How was the safety issue brought to light? What type of study did the FDA require to answer definitively the question about this serious risk? Can you determine whether this study was ever completed?
Let’s Go to the Videotape: Using Selected Clips from Webcast FDA Advisory Committees
You have probably heard the phrase ‘“Let’s go to the video” in televised sports news programs, when there is recorded videotape of a sports event and some segment is of interest to illustrate a performance or an issue of public interest to viewers. It is effective in highlighting a particular athletic event, a player’s performance, a controversial call.
This technique can also be used to illustrate how complex decision making can be in a multidisciplinary setting, in particular how evidence is evaluated in a regulatory setting. It also can illustrate the role of biostatisticians, clinical specialists, epidemiologists, and other scientists in providing guidance and insight into that evaluation.
The course we have developed at the Harvard T. Chan School of Public Health is based in part on the use of video clips, drawing substantially from FDA’s wealth of publicly available information, to illustrate case studies and construct case studies that illustrate regulatory decision making in action. Later in this article, we show how others can construct courses using this concept, which shows how to access and extract information to develop cases.
Case Study Illustrating the Impact of Missing Data on Evaluating Strength of Evidence
This case study includes information extracted from the FDA website as well as several video clips from an FDA advisory committee. LaVange and Permutt (2016) discuss this case study and the issues associated with missing data in clinical trials, including using imputation and unrealistic models that assume the dropout patterns are missing at random.
Missing data in clinical trials is one of the current topics of interest to the FDA and the clinical trial community. This case study illustrates how the strength of evidence of a clinical trial package is evaluated in the face of missing data, how the approval of a new drug is affected when data are missing, and how it is treated statistically in analysis and interpretation.
The topic has been discussed in the statistical literature for many years and methodology has been developed to address missing data, but most of the statistical formulations of the problem have been either unrealistic for clinical trials or not very helpful in interpreting clinical trial results and in making causal inferences about whether a drug is effective for its intended claims. The report “The Prevention and Treatment of Missing Data in Clinical Trials” (2010) developed by the National Research Council of the National Academies in response to an FDA request, and several FDA-authored articles on the topic (Permutt, 2016) provide useful background information.
The case study begins with the January 30, 2013, meeting of the FDA Pulmonary-Allergy Drugs Advisory Committee (PADAC) to consider a drug—mannitol inhalation powder—for managing cystic fibrosis in patients aged 6 and older and improve pulmonary function. (While the content and issues discussed are important, the focus of this article is on how to use the information in a classroom setting, or potentially in an online/on-demand situation.)
Clicking on the link opens a webpage that includes all the information about the meeting, including the agenda, committee roster, questions to the committee, FDA briefing materials made available to advisory committee members and the public, FDA and sponsor presentations (usually in PowerPoint slide format), transcript, and minutes of the meeting. Of interest to our case study is that this PADAC meeting was webcast. We treat it as the main vehicle to demonstrate how it can be used for training and education purposes.
After clicking on the link for the webcast, you see a screen that shows that the meeting recording is segmented in time slots. (Click image to enlarge)

Clicking on the Adobe Connect link (collaboration.fda.gov) for the segment on the start of the meeting through the morning break, for instance, provides a screenshot:
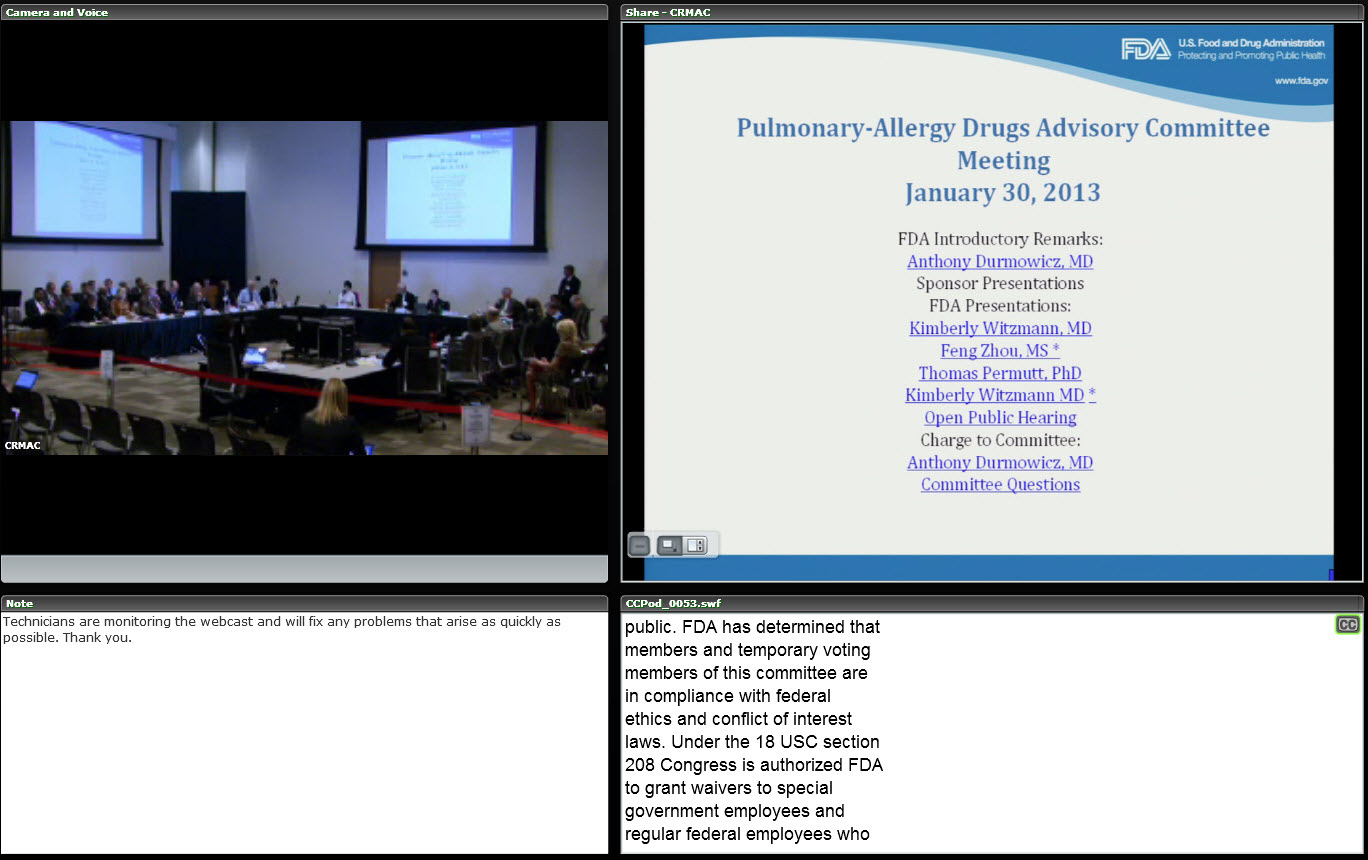
This screenshot shows how the committee table is structured, with the slides to be presented on the right of the screen and the time signature of the video on the bottom of screen. Various options are available to eliminate the text at the bottom or the relative size of the committee view and the projected slides.
Since the meeting is almost eight hours long, the webcast videos are separated into four segments. In this case study, we use eight clips, but it is possible to use fewer or more clips, depending upon how much material is to be covered. For in-class purposes, there is a time limit for how much can be shown and discussed in a single lecture, so one consideration for the design of each lecture is to assign students to view video clips before class, and then use class time to discuss the content.
For this case study, each of the four video segments has a time signature that can be used to select the portion of the video that is relevant to the instructional goal of a lecture. We use the following clips, which are indexed by their time signatures.
- Clip #1: Introduction and background of the issues presented by the FDA clinical division director, setting the framework for what the committee will be asked to discuss and vote upon. It is from the segment “Start of meeting to Morning Break” and is approximately 7.5 minutes long; time signature [0:8:00 to 0:15:20].
- Clip #2: From the segment “Morning Break to Lunch Break”; contains the presentation by FDA statistical reviewer Dr. Feng Zhou, about her evaluation of the evidence for efficacy of the product, the impact of missing data, and sensitivity analysis and patient subgroups. About 26 minutes [0:09:00 to 0:35:57].
- Clip #3: From the same segment as #2; contains comments from Thomas Permutt, MD, division director, on a different way of considering the missing data problem that does not rely upon modeling and assumptions about random missingness, and how to describe the treatment effect. About 11 minutes [0:36:24 to 0:47:44].
- Clip #4: From the segment “Lunch Break to Afternoon Break”; contains committee discussion of questions they were asked to address. Question 1 is about the evidence for efficacy and includes the division director’s instructions to the committee on the criteria for “substantial evidence” and for the balance of evidence [1:07:00 to 1:09:36]. About 40 minutes [0:52:00 to 1:32:23].
- Clip #5: From the same segment as #4; contains committee discussion of the safety profile of the drug, including a minor discussion of why the efficacy results appear different for a foreign subset of data in Argentina. About 40 minutes [1:32:23 to 2:14:52].
The next three clips present the critical voting questions that are based upon the questions the FDA asks the committee to vote on, about evidence and whether the evidence satisfies scientific regulatory standards.
- Clip #6: On question 4, about efficacy and the committee members’ reasons for their votes. About 7.5 minutes [0:01:30 to 0:9:00].
- Clip #7: A vote on question 5, about safety and the committee members’ reasons for their votes. About 6 minutes [0:09:00 to 0:15:00].
- Clip #8: A vote on question 6, about whether the efficacy and safety data provide substantial evidence for approval. About 5 minutes [0:15:15 to 0:20:23].
We do not always use Clips # 4 and 5 in class because of the time involved.
Case Study Illustrating Impact of Missing Data and Retrospective Attempts to Address It by Statistical Analysis Methods
In this case study, the concerns are about the role of missing outcome data for a single large clinical trial and its impact on a decision for approval of a new indication. It is based upon the deliberations at a January 16, 2014, FDA Cardiovascular and Renal Drugs Advisory Committee (CDRAC).
Topic: Discussion of the drug rivaroxabam (Xarelto® .5 mg) to reduce the risk of thrombotic cardiovascular events in patients in the first 90 days after suffering acute coronary syndrome (ACS)
Background: The drug Xarelto has been approved and is on the market for several indications, and the sponsor was seeking a new indication based upon a single clinical trial called ATLAS that was published in a medical journal (Mega, Braunwald, Wiviott, et al., 2012). The trial met its primary endpoint, but the FDA concern that was brought to the committee for discussion and a vote related to a retrospective, data-driven approach to dealing with missing outcome data that had a negative impact on the strength of evidence to support the claim made by the sponsor.
This is an example of the use of composite endpoints in a clinical trial where one of the components of the composite endpoint—cardiovascular death—was not known (not captured) for a substantial proportion of the randomized subjects because the subjects were not followed after they stopped taking the drug, but before the planned end of the trial.
The issue is about informative censoring. The magnitude of the treatment effect on mortality was much smaller than the magnitude of the missing data percentage, and looked to be differential among the treatment groups, with more missing outcome data apparent earlier in the trial course raising the potential for bias in the resulting comparisons.
This study followed 15,526 patients for a total of 16,820 person-years (mean follow-up of 13 months and up to 31 months) and looked at where the events could occur throughout the entire exposure and follow-up time.
To resolve the missing data concern, a post-hoc retrospective change in the analysis plan was suggested, along with shortening the time frame for counting when the outcomes occurred by using a 90-day window for measuring the occurrence of the endpoint to avoid missing data occurring at later times due to lack of patient follow-up. The questions to the committee involved whether this was a hypothesis-generating analysis or a confirmatory analysis.
Here is what students were expected to learn from this case study, especially from the video clips and the FDA briefing package to committee members.
- How FDA and its independent advisory committee evaluate the strength of evidence provided by a clinical study, even one that was peer-reviewed and published in the medical literature, and how the claim (indication) sought by the sponsor is evaluated or modified.
- Why two previous CRDAC meetings that evaluated the same study evidence voted against approval for the sought indication, and why new analyses of the same study data were conducted to both address missing data and optimize the benefit/risk profile of the drug relative to the duration of patient exposure to the drug and use of the drug.
- The ultimate impact of missing outcome data and other potential biases that weaken the strength of evidence when only a single study is available to support a claim of substantial evidence. A reading of the published article alone would not provide the information needed to understand why the advisory committee did not recommend approval for this indication. The video clips in this case study help viewers understand the depth, thoroughness, science, and concerns behind the FDA’s review and decision making.
View and download all meetings materials, here.
To fully understand what occurred at the meeting and the various discussions, presentations by FDA, and sponsor and the committee deliberations, view the archived video of the entire one-day advisory committee meeting.
We selected only certain clips from the all-day meeting that we think convey the issue:
- Introductory remarks by the Cardiorenal Drugs division director, Dr. Norman Stockbridge, regarding why the topic was brought to the committee and what the committee is being requested to consider (3.5 minutes; [00:07:09 to 0:10:11]).
- Discussion of Question 5 regarding impact of missing data and strength of evidence (23 minutes; [0:02:26 to 0:25:20]).
- Voting on the question of whether the drug should be approved for the requested indication (17 minutes; [0:26:00 to 0:43:40]).
How to Develop a Case Study Using Publicly Available FDA Material
Template for a case study
1. Choose a theme or a topic that can be linked to a methodological topic in a related course, such as clinical trials, epidemiology, pharmacology or drug development, active surveillance, meta-analysis, etc. Generally, the case study will reflect the scientific and regulatory evaluation of evidence of efficacy and safety or risk/benefit but in the context of a specific drug/disease area. The case study is specifically designed to use publicly available information on FDA’s website and, in particular, video from FDA advisory committees that has been webcast and archived in the FDA advisory committee website.
2. Structure the case study so the lecturer or preparer develops all the materials for the case according to the messages and lessons to be learned, and leads the students in active discussion and feedback. It also can be structured to direct students/participants to develop the case study themselves with the advice or direction of a lecturer and by using various FDA information resources.
- 3. Use FDA information resources available to create the case study:
a. FDA advisory committee website, which contains archived materials from each advisory committee, many of which are webcast; indexed by calendar year, date of meeting, and therapeutic area. Generally, the following information is available for downloading: meeting announcement and agenda, roster of members, questions to the committee, briefing materials, sponsor and FDA presentations (PowerPoint slides or PDF), transcript, webcast video, and meeting minutes.
Since most meetings are at least eight hours in length and may last over two days, there is a considerable amount of material to digest. The challenge to the preparer in creating a case study is to select materials that focus most closely on the topic and lessons to be learned. The meeting materials can be viewed as reading assignments or in various combinations of in-class and online access.
b. If a drug has been approved, the record of its approval, including the written reviews of each subject-matter FDA reviewer and the decision memos of division directors or office directors. This is available at Drugs@FDA and downloadable, accessible by drug name. The written reviews can be quite lengthy and might serve as selective reading assignments for students responsible for developing case studies.
c. To evaluate whether the evidence submitted by a sponsor in a New Drug Application or Biologics Application meets the scientific evidentiary standards for substantial evidence of efficacy —a major role and responsibility of the FDA. This process is generally poorly understood by the public; one of the few opportunities for the public to witness and learn about the breadth of evidence evaluation is through the public advisory committees.
Most of the topics brought to an FDA advisory committee deal with the evidence for the safety, efficacy, or benefit/risk of a new drug or biologic. The structure of the meeting involves asking the committee questions about whether the sponsor’s data have met the evidentiary standards for efficacy and safety. Knowledge of the evidentiary standards is important as both background and a basis for evaluation. This information is available in the Code of Federal Regulations and often summarized by FDA officials at advisory committee meetings.
d.Published guidances for industry that provide scientific guidance and direction on clinical trial study design, endpoint selection, good practice and procedures, therapeutic specific advice, use and interpretation of foreign clinical data, etc: http://bit.ly/2hwUCEN. These guidances are another possible resource for case-study preparers.
Further Reading
Krantz, M.J., and S. Kaul. 2013. The ATLAS ACS-TIMI 51 Trial and the Burden of Missing Data. Journal of the American College of Cardiology 62(9), 777–781.
LaVange, L.M., and T. Permutt. 2016. A regulatory perspective on missing data in the aftermath of the NRC report. Statistics in Medicine 35(17), 2,853–64.
Little, R.J., J. Wang, X. Sun, H. Tian, et al. 2016. The treatment of missing data in a large cardiovascular clinical outcomes study. Clinical Trials 13(3), 344–351.
Mega, J.L., E. Braunwald, S.D. Wiviott, et al., for ATLAS ACS-TIMI 51 investigators. 2012. Rivaroxaban in Patients with a Recent Acute Coronary Syndrome. New England Journal of Medicine 366, 9–19.
National Research Council of the National Academies. 2010. The Prevention and Treatment of Missing Data in Clinical Trials. Washington, DC: National Academies Press.
O’Neill, R.T. 2006. FDA’s critical path initiative: A perspective on contributions of biostatistics. Biometrical Journal 48, 559–564.
Permutt, T. 2016. For the Missing Data Working Group. Sensitivity analysis for missing data in regulatory submission. Statistics in Medicine 30;35(17), 2,876–9.
Permutt, T. 2016. For the Missing Data Working Group. A taxonomy of estimands for regulatory clinical trials with discontinuations. Statistics in Medicine 30;35(17), 2,865–75.
Proschan, Michael. 2016. FDA Advisory Committees: The Role of Statisticians. CHANCE 29(4), 31–40.
U.S. Food and Drug Administration. 2006. Innovation/Stagnation–—Challenge and Opportunity on the Critical Path to New Medical Products. Washington, DC: FDA.
U.S. Food and Drug Administration. 2011. Advancing Regulatory Science for Public Health. Washington, DC: FDA.
U.S. Food and Drug Administration. 2016 (updates). FDA’s Sentinel System. Washington, DC: FDA.
U.S. Institute of Medicine, National Academies of Sciences, Engineering, and Medicine. 2011. Strengthening a Workforce for Innovative Regulatory Science in Therapeutics Development–Workshop Summary. Washington, DC: National Academies Press.
This article reflects the views of the author and should not be construed as representing views or policies of the U.S. Food and Drug Administration.
About the Authors
Robert T. O’Neill is the senior statistical advisor and formerly director of the Office of Biostatistics at the Center for Drug Evaluation and Research (CDER), FDA. His office oversaw the statistical review of submitted drug applications and was responsible for setting statistical policy. O’Neill was the ICH FDA topics leader for two guidances, E9 and E5. He is a Fellow of the ASA and the Society for Clinical Trials, and the 2002 recipient of the Marvin Zelen Leadership Award in Statistical Science. O’Neill recently developed a course entitled “Statistical and Quantitative Methods for Pharmaceutical Regulatory Science” at the Harvard T.H. Chan School of Public Health.
Marcia A. Testa is a senior lecturer on biostatistics at the Harvard T.H. Chan School of Public Health. Testa and colleagues are developing quantitative and health information technology methods and processes relating to the measurement and analysis of expanded patient outcomes (quality of life, cost-effectiveness, and risk-benefit) for clinical, pharmacoeconomic, pharmacoepidemiologic, quality of care, and comparative effectiveness research studies with particular application to primary care, cardiovascular disease, HIV, and diabetes.
Year 2014 is expected to bring new awareness in users.Retaining today’s
multigeneational workforce also typically involves establishing a cohesiv social media strategy.
If we balance thhe amazing technologies tbat we have and all
the world of information they hold with personal communication and hard work, the resultys will be moire positive,
tthe accountability will show what it is supposed to show,
and students will leave our doors knowing more than when they came in.